Reference Manual
maketrnadb.py
maketrnadb.py creates the reference database that is used by processsamples.py for analyzing small RNA-seq data. It uses Bowtie2 to build indexes with tRNA sequences and reference genome sequence for read alignments. It also uses Infernal to create tRNA sequence alignments for annotating the positions of sequencing reads and misincorporations. The tRNA annotations required as inputs can be downloaded from GtRNAdb or generated by running tRNAscan-SE. The reference genome sequence can be downloaded from the UCSC Genome Browser.
Usage
maketrnadb.py --databasename=dbname --genomefile=genome.fa
--trnascanfile=trnascan.txt [--gtrnafafile=db-tRNAs.fa --namemap=db-namemap.txt]
Options
- --databasename = dbname (required)
Directory and database name that will be used for the reference database - --genomefile = genome.fa (required)
FASTA file of reference genome - --trnascanfile = trnascan.txt (required)
tRNAscan-SE output file (*.out file in GtRNAdb downloaded tarball) - --gtrnafafile = db-tRNAs.fa (optional)
tRNA sequence FASTA file downloaded from GtRNAdb - --namemap = namemap.txt (optional)
Map file that converts tRNAscan-SE IDs to GtRNAdb gene symbols - --orgmode = mode (optional)
Sequence source of reference
Default is "euk" for eukaryotes. Other option includes "mito" for mitochondria, "arch" for archaea, and "bact" for bacteria.
(Archaeal and bacterial options are currently under development. The mito option may not work properly for degenerative mitochondrial tRNAs. We suggest downloading the pre-built reference databases with the use ofquickdb.bashinstead.) - --forcecca = mode (optional)
Force addition of 3' CCA to tRNA for bacteria and archaea. By default, 3' CCA will be added automatically to eukaryotic mature tRNAs.
Outputs
The following files are generated upon completion:
dbname-tRNAgenome.*: FASTA file of tRNA and reference genome sequences with Bowtie2 indexesdbname-trnaalign.stk: Alignments of mature tRNA isodecoder sequences in Stockholm file formatdbname-trnaloci.stk: Alignment of tRNA gene sequences in Stockholm file formatdbname-trnatable.txt: Tab-delimited file with tRNA isodecoders and tRNA genes mapdbname-maturetRNAs.fa: FASTA file of mature tRNA isodecoder sequencesdbname-maturetRNAs.bed: tRNA isodecoders in BED file formatdbname-trnaloci.bed: tRNA genes in BED file formatdbname-dbinfo.txt: Database creation information and logdbname-alignnum.txt: tRNA position numbering table for tRNAdbname-locusnum.txt: tRNA position numbering table for pre-tRNAs
trimadapters.py
trimadapters.py removes adapter sequences from raw sequencing data as the preprocessing step before data analysis. For single end reads, Cutadapt is used for performing the adapter trimming. For paired end reads, the two read ends have to be merged into single end reads in order to allow correct read alignments by Bowtie2. Therefore, SeqPrep is used that will trim adapter sequences and merge the read ends simultaneously. The tool was designed to work with raw sequencing data generated by Illumina platform and has not been tested with data from other sequencing platform.
Note
The options for supporting Unique Molecular Identifiers (UMI) are experimental and may only be applicable for specific designs. Researchers who are interested in utilizing the tool with sequencing data containing UMI can contact us for feasibility inquiries.
Usage
trimadapters.py --runname=runname --runfile=runfile.txt [--firadapter=FIRADAPTER --secadapter=SECADAPTER --minlength=MINLENGTH --singleend --cores=CORES]
Options
- --runname = runname (required)
Name used for output files - --runfile = runfile (required)
Tab or whitespace-delimited file with sample and FASTQ file information (described below) - --firadapter = FIRADAPTER (optional)
3' adapter sequence used in single end reads or forward primer/adapter sequence used in paired end reads
Default value is AGATCGGAAGAGCACACGTC - --secadapter = SECADAPTER (optional)
Reverse primer/adapter sequence used in paired end reads
Default value is GATCGTCGGACTGTAGAACTC - --minlength = MINLENGTH (optional)
Minimum length of sequencing reads to be retained after trimming and/or merging
Default value is 15 - --singleend
Flag to specify single end reads - --cores = CORES (optional)
Number of cores to be used
Default value is the number of available cores on the system - --umilength = length (optional)
Length of UMI
Default value is 0 - --umithreeprime (optional)
Flag to indicate that UMI is located at the 3' end of the reads instead of 5' end. It will have no effect if --umilength is 0.
Input files
Raw sequencing files
These files should be in FASTQ file format and can be compressed by gzip. They are specified in the runfile.
trimadapters.py runfile
This is a two or three-column tab-delimited text file depending on single or paired end reads. The columns are defined as:
- column 1: sample name
- column 2: single-end FASTQ file name or read 1 FASTQ file name for paired end reads
- column 3: read 2 FASTQ file name for paired end reads
The FASTQ file names may include the file paths where the files are located.
Outputs
The following files are always generated upon completion:
runname_log.txt: Output log containing statistics generated by Cutadapt or SeqPreprunname_manifest.txt: List of sample names and trimmed/merged FASTQ files
For single end reads, the following files are created:
samplename_trim.fastq.gz: FASTQ file containing trimmed sequencing reads and can be used by processsamples.pyrunname_ca.pdf: histogram summarizing the amount of trimmed, untrimmed, and discarded readsrunname_ca.txt: Data source for the histogram inrunname_ca.pdf
For paired end reads, the following files are created:
samplename_merged.fastq.gz: FASTQ file containing trimmed and merged sequencing reads and can be used by processsamples.pysamplename_left.fastq.gz: FASTQ file containing Read 1 that do not mergesamplename_right.fastq.gz: FASTQ file containing Read 2 that do not mergerunname_sp.pdf: histogram summarizing the amount of merged, unmerged, and discarded readsrunname_sp.txt: Data source for the histogram inrunname_sp.pdf
processamples.py
processamples.py is the main tool that performs analysis on the small RNA sequencing data. It includes aligning reads to the reference database using Bowtie2, estimating transcript abundance, performing differential expression comparison across samples, and detecting misincorporations as potential RNA modifications. Multiple outputs in text files and images are generated during different steps of the analysis. For details of the analysis methods used in processamples.py, please refer to the Methods section of our publication.
Usage
processsamples.py --experimentname=expname --databasename=dbname --ensemblgtf=genes.gtf.gz --samplefile=samplefile.txt [--exppairs=samplespairs.txt --bedfile=file.bed --minnontrnasize=SIZE --nofrag --lazyremap --makehub --olddeseq --dumpother]
Options
- --experimentname = expname (required)
Experiment name that will be used for results - --databasename = dbname (required)
Directory and name of reference database generated by maketrnadb.py - --ensemblgtf = genes.gtf.gz (required)
Non-tRNA annotation file in Ensembl GTF file format - --samplefile = samplefile.txt (required)
Tab or whitespace-delimited file with sample and pre-trimmed FASTQ file information (described below) - --exppairs = samplespairs.txt (optional but recommended)
Tab or whitespace-delimited file with sample pairs for differential expression comparison (described below)
If not provided, all vs all pairwise comparison will be performed. - --bedfile = file.bed (optional)
Other non-tRNA gene annotations in BED file format
Multiple files can be specified. - --minnontrnasize = SIZE (optional)
Minimum read length to be retained for non-tRNA genes
Defaults value is 20 - --nofrag
Flag to skip tRNA fragment determination
Design for full length tRNA sequencing experiments - --lazyremap
Skip read mapping step when BAM files already exist - --makehub
Convert BAM files by interpolating tRNA read alignments to the reference genome and create read coverage tracks in track hub to be used in UCSC Genome Browser - --olddeseq
Use DESeq1 instead of DESeq2 - --dumpother
Output read alignments that do not have an associated gene feature into BAM files (files end in "nofeat.bam") - --cores
Number of processing cores to be used (default = 8)
Input files
Trimmed sequencing files
These files should be in FASTQ file format and can be compressed by gzip. They are specified in the samplefile. Adaptor sequences should have been removed from the sequencing reads. Only single end reads are supported. If paired end reads are used in sequencing, the two read ends that always overlap in small RNA-seq should be merged into single end reads. These files can be output files generated by trimadapters.py or other preferred tools.
Sample File
samplefile is a three-column tab-delimited file of sample meta data. The columns are:
- column 1: unique sample replicate name
- column 2: sample or group name common for replicates
- column 3: preprocessed FASTQ file name
An example sample file TestSamples.txt (used for TestRun.bash) can be found with the tRAX source code.
Sample pair file
exppairs is a two-column tab-delimited file consists of sample pairs to be compared with each other. The columns are:
- column 1: sample name
- column 2: another sample name
The sample names must match with column 2 values in the sample file above.
Outputs
The results generated by processamples.py are distributed into multiple subfolders, outlined as follows:
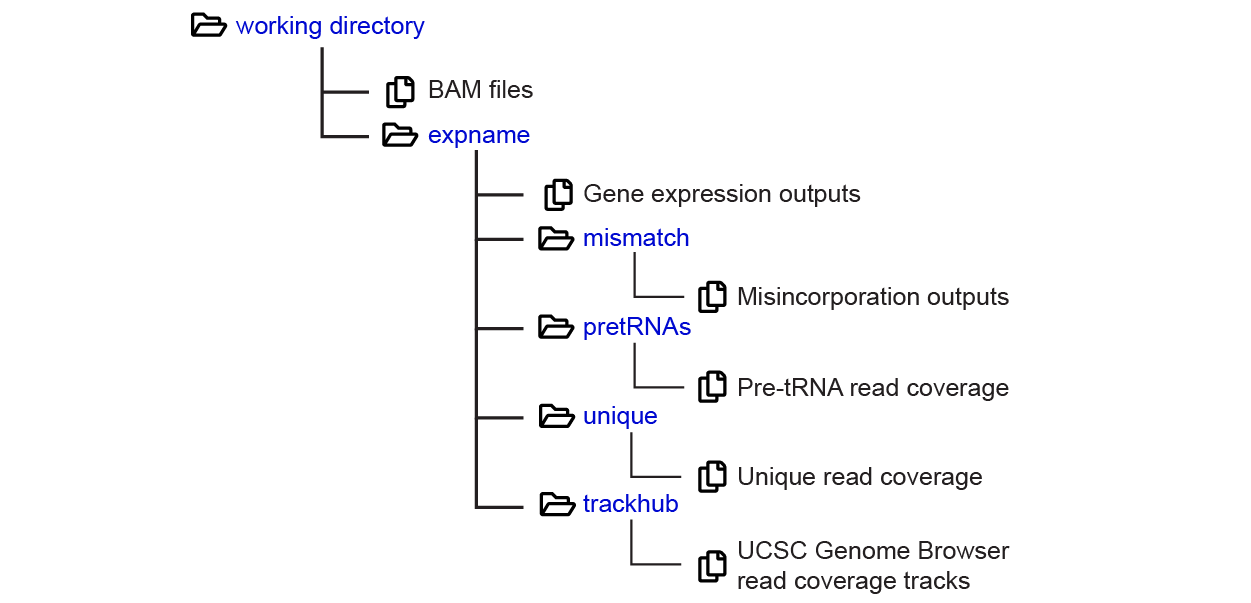
Analysis run info
expname-runinfo.txt: Information on the tRAX run, including input parameters, run date, run command, and tRAX version
Quality assessment report
expname-qa.html: HTML document showing quality assesment of sequencing reads and alignments
Read alignments
sample_name.bam: Sorted BAM file with read alignments ofsample_namesample_name.bam.bai: BAM index file ofsample_name.bamsample_name-genome.bam: BAM file with read alignments ofsample_namemapped to reference genome coordinates, used for UCSC Genome Browser track creationsample_name_nofeat.bam: BAM file without gene features, generated when using--dumpotheroptionexpname-mapstats.txt: Bowtie2 command and raw output statistics for each sampleexpname-mapinfo.txt: Mapping statistics for each sample, used as input forexpname-mapinfo.pdfexpname-mapinfo.pdf: Histogram of mapping rate for each sample
Summary of read coverage statistics
expname-typecounts.txt: Normalized read counts of small RNA types per sampleexpname-typecounts.pdf: Normalized read distribution of small RNA types per sampleexpname-typerealcounts.txt: Raw read counts of small RNA types per sampleexpname-typerealcounts.pdf: Raw read distribution of small RNA types per sampleexpname-aminocounts.txt: Normalized read counts of tRNA isotypes per sampleexpname-aminocounts.pdf: Normalized read distribution of tRNA isotypes per sampleexpname-combinedcoverage.pdf: Read coverage summary of tRNA isotypes per sampleexpname-readlengths.txt: Counts of read lengths for tRNAs, pre-tRNAs, and other gene types per sampleexpname-readlengths.pdf: Distribution of read lengths for tRNAs, pre-tRNAs, and other gene types per sample
Abundance of tRNA, tDRs, and other gene types
sample1_sample2-typescatter.pdf: Sample pair comparisons of normalized read counts for each gene type, including tRNA fragment type when--nofragis not usedsample1_sample2-aminoscatter.pdf: Sample pair comparisons of normalized read counts for each tRNA fragment type when--nofragis not used or for tRNAs when--nofragis usedexpname-readcounts.txt: Raw read counts for each transcript and tRNA fragmentexpname-normalizedreadcounts.txt: Normalized read counts for each transcript and tRNA fragmentexpname-coverage.txt: Per-base read coverage data of all tRNA isodecoders per sample, using Sprinzl tRNA positioningexpname-coverage.pdf: Per-base read coverage plot of all tRNA isodecoders per sample colored by specificity of mappingexpname-<isotype>_cov.pdf: Individual isotype per-base read coverage plots of tRNA isodecoders per sample colored by specificity of mappingexpname-SizeFactors.txt: DESeq2 size factors for each sampleexpname-trnaendcounts.txt: Counts of tRNA "CCA" tail types
Differential expression comparison
expname-logvals.txt: Log2 fold change values of sample pairs generated by DESeq2expname-padjs.txt: Adjusted P-values of sample pairs generated by DESeq2expname-combine.txt: Tab-delimited file including adjusted P-values, log2 fold change values, and normalized read countssample1_sample2-volcano.pdf: Volcano plots of sample pair with log2 fold change values and adjusted P-values Top 10 results are labeledsample1_sample2-volcano_tRNA.pdf: Volcano plots of sample pair with log2 fold change values and adjusted P-values of only tRNAs and tRNA fragments. Top 10 results are labeledexpname-pca.pdf: Principle component analysis of samples, based on normalized read counts of all gene featuresexpname-pcatrna.pdf: Principle component analysis of samples, based on normalized read counts of tRNAs and tDRs
Base mismatch estimation
mismatch/expname-trnapositionmismatches.pdf: Maximum mismimatch percentage among all samples for each tRNA positionmismatch/expname-trnapositionfiveprime.pdf: Maximum percentage of start position among all samples for each tRNA positionmismatch/expname-<isotype>_mismatch.pdf: Per-base coverage of mismatches for tRNA isodecodersmismatch/expname-<isotype>_delete.pdf: Per-base coverage of deletion for tRNA isodecodersmismatch/expname-<isotype>_fiveprimeends.pdf: Per-base coverage of 5' start position for tRNA isodecodersmismatch/expname-<isotype>_mismatchheatmap.pdf: Heatmap of mismatches per base for tRNA isodecoders of specific isotypemismatch/expname-<isotype>_deletionheatmap.pdf: Heatmap of deletions per base for tRNA isodecoders of specific isotypemismatch/expname-<isotype>_threeprimeheatmap.pdf: Heatmap of 3' end position for tRNA isodecoders of specific isotypemismatch/expname-<isotype>_fiveprimeheatmap.pdf: Heatmap of 5' start postion for tRNA isodecoders of specific isotypemismatch/expname-<isotype>_fiveprimecounts.pdf: Distribution of 5' start position of all tRNA transcripts for each samplemismatch/expname-<isotype>_threeprimecounts.pdf: Distribution of 3' end position of all tRNA transcripts for each samplemismatch/expname-<isotype>_trnapositionmismatches.pdf: Maximum mismimatch percentage among all samples for each tRNA position by anticodonmismatch/expname-<position>_posaminomismatches.pdf: Mismatch percentage at position per tRNA isotypemismatch/expname-<position>_possamplemismatches.pdf: Mismatch percentage at position per samplemismatch/expname-<position>_posaminodeletions.pdfs: Deletion percentage at position per tRNA isotypemismatch/expname-<position>_possampledeletions.pdf: Deletion percentage at position per samplemismatch/expname-<position>_possamplereadstarts.pdf: Read start percentage at position per sample
Pre-tRNA read coverage
pretRNAs/expname-pretRNAcombinedcoverage.pdf: Read coverage summary of tRNA isotypes per sample, with upstream and downstream regionspretRNAs/expname-pretRNAcoverage.txt: Per-base read coverage data of all tRNA isodecoders per sample, with upstream and downstream regionspretRNAs/expname-pretRNAcoverage.pdf: Per-base read coverage plot of all tRNA isodecoders per sample, with upstream and downstream regions
Unique read coverage
unique/expname-trnauniquecounts.txt: Counts of uniquely mapped reads for all transcripts per sampleunique/expname-<isotype>_uniqueonlycov.pdf: Individual isotype per-base unique read coverage plots of tRNA isodecoders per sample
Genome browser track hub
trackhub/<sample>.Plus.bw: Read coverage BigWig file of specific sample on forward strandtrackhub/<sample>.Minus.bw: Read coverage BigWig file of specific sample on reverse strandtrackhub/trackdb.txt: UCSC Genome Browser track information in track database definition format
gettranscripts.bash
gettranscripts.bash is a tool for generating transcript predictions from a bam file. It is intended to be used on the output from the --dumpother option of processamples.py after merging with samtools. This requires bedtools to be installed to function
Usage
gettranscripts.bash mergedoutput.bam 30 > output.bed
Arguments
- First argument is the bam file generated by processamples.py with
--dumpotheroption. - Second argument is the minimum number of transcripts.
Output
output.bed: BED file with possible unannotated transcripts found in the BAM file