Getting Started
About
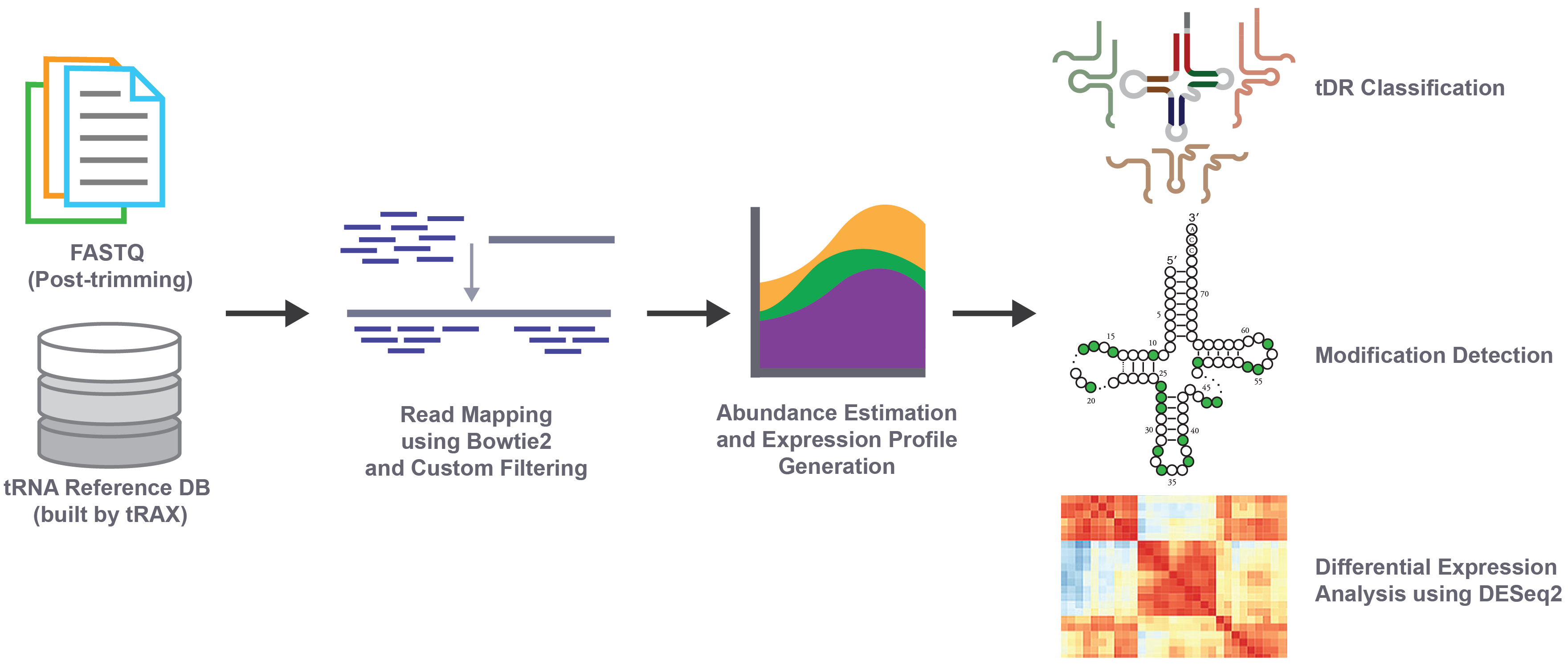
tRNA Analysis of eXpression (tRAX) is a software package built for in-depth analyses of tRNA-derived small RNAs (tDRs), mature tRNAs, and inference of RNA modifications from high-throughput small RNA sequencing data. While the tRAX workflow adopts popular RNA sequencing data analysis methods, which includes adapter trimming of raw sequencing data, read alignment to the reference, transcript abundance estimation, and differential expression analysis among samples, it specifically consists of features designed to support special characteristics of tRNAs and tDRs. To ensure alignment of sequencing reads to tRNA transcripts, tRAX uses a custom-built reference database that not only includes the reference genome but also mature tRNA transcripts with the addition of 3′ CCA tail not encoded genomically. Unlike popular read counting tools that only consider or recommend for uniquely mapped reads for RNA sequencing, tRAX allows reads to be mapped to multiple transcripts and gene loci, which is necessary for conserved tRNA isodecoders (tRNAs with the same anticodon but different sequences in the gene body) and identical tRNA genes that are commonly found in eukayotes. Read coverage for each tRNA transcript is reported in four categories – transcript-specific, isodecoder-specific, isotype-specific, and non-specific – that provide precise results on the level of uniqueness of the aligned reads. Moreover, tRAX computes separate read counts of tRNA fragments that align at the 5′ end, 3′ end, and the middle region of tRNA transcripts to distinguish the abundance of different fragment types. Differential expression comparison across samples is performed using read counts for tRNA transcripts and tRNA fragments to provide better understanding of possible distinction in different tRNA isotypes or isodecoders and fragment types. In addition, tRAX measures the base frequency at each position aligned to the tRNA transcripts for estimating the mis-incorporations that may represent RNA modifications essential for function, stability, and regulation.
To enable researchers to study the analysis results at multiple levels, tRAX presents over 50 types of visual images and data files such as read distribution summarizations, read coverage per tRNA isodecoder, volcano plots of differential expression comparison between samples, and misincorporation location charts. More details are available at Understanding Output Results and the Reference Manual.
Installation
System requirements
tRAX requires to be run on a Linux/Unix system with at least 8 cores and 16 GB memory. Due to the large size of sequencing data, we do not recommend using tRAX on a regular desktop or laptop.
Using Docker Image
To eliminate the need of installing dependencies, you can download the Docker image from our DockerHub repository using the command
docker pull ucsclowelab/trax
Using Conda Environment
For those who prefer to use conda, you can create the environment using the command
conda env create -f trax_env.yaml
Getting source code
The source code can be downloaded from GitHub at https://github.com/UCSC-LoweLab/tRAX. tRAX was developed with Python and does not require compilation or installation.
To run tRAX from source code, dependencies listed below are required to be installed.
Dependencies
- Python 2.7 or higher
- pysam Python library (latest verion - older versions have a memory leak)
- Bowtie2
- Samtools 1.9 or higher
- R
- DESeq2 bioconductor library 1.26 or higher (DESeq a possible substitution)
- getopt R library
- ggplot2 R library
- ggrepel R library
- Infernal 1.1.2 or higher
- Cutadapt 1.18 (required for running trimadapters.py)
- SeqPrep (required for running trimadapters.py)
- NCBI SRA toolkit (required for test run)
Tutorial
Test run
To try out tRAX with a small public data set, we provide a script TestRun.bash. It includes downloading small RNA sequencing data from NCBI SRA, trimming sequencing adapters, building the reference database using the human tRNA annotations from GtRNAdb and the GRCh37/hg19 reference genome from UCSC Genome Browser, and analyzing the data with results generation. The script should be run in the directory where outputs are desired to be stored.
Additionally, ARM-Seq and DM-tRNA-seq data described in the following publications can be downloaded at http://trna.ucsc.edu/tRAX/data/ARMseq and http://trna.ucsc.edu/tRAX/data/DMtRNAseq respectively. Sample meta data files are also provided. Please refer to the included README for description and usage.
Cite
Cozen AE, Quartley E, Holmes AD, et al. (2015) ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nature Methods 12:879–884.
Zheng G, Qin Y, Clark WC, et al. (2015) Efficient and quantitative high-throughput tRNA sequencing. Nature Methods 12:835–837.
Components
tRAX contains three main tools:
maketrnadb.py- Build a reference database for sequencing data analysistrimadapters.py- Trim adapters from raw sequencing readsprocessamples.py- Analyze small RNA-seq data
Data needed as inputs
- Raw sequencing data from Illumina platform in FASTQ file format
- tRNAscan-SE outputs of targeted genome downloaded from GtRNAdb
- Genome sequence FASTA file (can be downloaded from UCSC Genome Browser)
- Non-tRNA gene annotations in Ensembl GTF file format
Note
The chromosome names must be the same across all the input files. For example, if chr1 is used as chromosome 1 in tRNA annotations, the same chromosome name must be used in the genome sequence FASTA file and the non-tRNA gene annotation GTF file.
Info
If the GTF file is downloaded from Ensembl, the following command can be used as an example for converting the chromosome names to UCSC Genome Browser style and obtaining the non-coding RNA annotations.
wget -q -O - ftp://ftp.ensembl.org/pub/release-75/gtf/homo_sapiens/Homo_sapiens.GRCh37.75.gtf.gz | gzip -cd | grep -v '^#' | awk '{print "chr" $0;}' | grep -e Mt_rRNA -e miRNA -e misc_RNA -e rRNA -e snRNA -e snoRNA -e ribozyme -e sRNA -e scaRNA > hg19-genes.gtf
How to Run
Step 1: Build custom reference database
Before analyzing the sequencing data, a custom reference database has to be built. If tRAX is used with human GRCh37/hg19, human GRCh38/hg38, mouse GRCm38/mm10, or Saccharomyces cerevisiae S288c, you can use the provided script quickdb.bash that automatically downloads the required input files and builds the database. The command is:
quickdb.bash <genome> <path (optional)>
<genome> = hg19 for human GRCh37/hg19or
hg38 for human GRCh38/hg38or
mm10 for mouse GRCm38/mm10or
sacCer3 for Saccharomyces cerevisiae S288c
For mitochondrial tRNAs, this command downloads the mito tRNA database without the need of using maketrnadb.py as described below. (The current version of maketrnadb.py does not work properly for degenerative mitochondrial tRNAs.)
or hg19mito for human GRCh37/hg19 mito tRNAs
or hg38mito for human GRCh38/hg38 mito tRNAs
or mm10mito for mouse GRCm38/mm10 mito tRNAs
By default the reference database will be created in the same folder the command is run unless an optional path is specified as a secondary argument.
If using a docker container the command, quickstart.bash build <genome> can be used to generate a docker volume containg the database that is mounted at /rnadb of the docker container. /rnadb will be the container directory for mounting volume when running docker container.
If another directory is desired, it can be supplied as the second argument when running quickdb.bash.
The reference database can also be built using maketrnadb.py with the pre-downloaded input files.
maketrnadb.py --databasename=dbname --genomefile=genome.fa
--trnascanfile=trnascan.txt --namemap=db-tRNAs_name_map.txt
dbnameis the name that will be used for the reference databasegenome.facontains a FASTA file of the reference genometrnascan.txtis the output file generated by tRNAscan-SE and can be downloaded from GtRNAdbdb-tRNAs_name_map.txtis the map file that converts the tRNAscan-SE IDs to GtRNAdb gene symbols. It is also included in the GtRNAdb downloaded tarball.
If maketrnadb.py is run within the tRAX source directory, a file path where the reference database will be created should be included as part of the dbname, for example, /db_path/hg38.
Step 2: Remove sequencing adapters
Raw sequencing data has to be preprocessed before analysis. This includes removing sequencing adapters and merging paired end reads into single end reads.
A tab or whitespace delimited file with sample information has to be prepared as input for trimadapters.py. The file contains two or three columns depending on single or paired end reads.
- column 1: sample name
- column 2: single-end FASTQ file or read 1 FASTQ file for paired end reads
- column 3: read 2 FASTQ file for paired end reads
Example
For single end reads:
sample1 samplefile1.fq.gz
sample2 samplefile2.fq.gz
sample3 samplefile3.fq.gz
for paired end reads:
sample1 samplefile1_1.fq.gz samplefile1_2.fq.gz
sample2 samplefile2_1.fq.gz samplefile2_2.fq.gz
sample3 samplefile3_1.fq.gz samplefile3_2.fq.gz
To preprocess the single end sequencing data, run
trimadapters.py --runname runname --runfile runfile.txt --singleend
trimadapters.py --runname runname --runfile runfile.txt
runnameis the name to be used in output filesrunfile.txtis the sample information file described above
The tool should be run in the directory where the output files will be generated. The output FASTQ file with trimmed sequencing reads will be named as *_trimmed.fastq.gz for single end reads or *_merge.fastq.gz for paired end reads.
Step 3: Analyze sequencing data for gene expression
Two meta data files have to be created as inputs for analysis. The first one is a tab or whitespace delimited sample file with three columns:
- column 1: unique sample replicate name
- column 2: sample or group name common for replicates
- column 3: preprocessed FASTQ file name
Example
WildType1 WildType samplefile1_merged.fq.gz
WildType2 WildType samplefile2_merged.fq.gz
WildType2 WildType samplefile3_merged.fq.gz
Heterozygous1 Heterozygous samplefile4_merged.fq.gz
Heterozygous2 Heterozygous samplefile5_merged.fq.gz
Heterozygous3 Heterozygous samplefile6_merged.fq.gz
Homozygous1 Homozygous samplefile7_merged.fq.gz
Homozygous2 Homozygous samplefile8_merged.fq.gz
Homozygous3 Homozygous samplefile9_merged.fq.gz
The second meta data file contains the pairs of samples for differential expression comparison. The sample names must match with the sample/group names (column 2) of the above sample file.
Example
WildType Heterozygous
WildType Homozygous
To start the analysis for mature tRNAs, run the following command:
processsamples.py --experimentname=expname --databasename=dbname
--ensemblgtf=genes.gtf.gz --samplefile=samplefile.txt
--exppairs=samplespairs.txt --nofrag
For tRNA-derived small RNAs, omit the --nofrag option. If desired to generate UCSC Genome Browser read coverage tracks for visualization, include the --makehub option.
expnameis a name that will be used for results as well as the folder that contains the resultsdbnameis the directory and name of the reference database generated bymaketrnadb.pygenes.gtf.gzis the non-tRNA gene annotation GTF filesamplefile.txtis the sample file described abovesamplespairs.txtis the sample pair file described above
The BAM files containing the read alignments will be saved in the current working directory while the other output results will be located in the sub-directory named as expname.